

为探究这些复杂而精密的分子机器的结构,在过去几十年中,以X射线晶体学(X-ray crystallography)、核磁共振波谱学(nuclear magnetic resonance spectroscopy, NMR) 和冷冻电镜(cryo-electron microscopy, Cryo-EM)为代表的解析技术应运而生,并获得了一系列重要发现[7-8]。作为奠基性技术,X射线晶体学带来了DNA双螺旋、血红蛋白等无数生物大分子的原子分辨率图像,构筑了分子生物学的理论基石[9-10]。核磁共振波谱学则独辟蹊径,在溶液等近生理条件下捕捉分子的动态变化和柔性区域信息,极大丰富了人们对蛋白质功能的认知[11-12]。而近年来冷冻电镜更是依靠“分辨率革命”成为结构解析的主流方法,克服了传统方法难以企及的复合物结构解析等挑战[13-15]。这些技术的发展,共同绘制了生命活动的分子蓝图,极大推动了现代生命科学的发展。
本文聚焦于生物大分子结构数据库,系统梳理其从经典存档体系到AI时代核心基座的演进路径。首先,描述PDB等核心生物大分子结构数据库的发展历程与数据生态,揭示其作为结构生物学研究基础设施的奠基性价值;进而阐述这些数据库如何为AlphaFold等AI模型在结构预测、功能注释等领域的突破提供关键的数据养料和知识前提;接下来深入探讨AI预测结果反哺并与实验数据结合后,所形成的“实验-计算”协同发展新范式;最后,基于这一新范式,展望未来智能化生物大分子结构数据库的架构趋势,及其在推动生命科学自动化发现进程中的巨大潜力。
1 生物大分子结构数据库体系及演进路径
生物大分子结构数据库的发展与结构生物学研究进程紧密相伴,是该领域不可或缺的基础设施。从作为基石的PDB数据库开始,数据库体系逐渐演化,催生了如SCOP(structural classification of proteins)等结构分类数据库,专注于冷冻电镜研究的EMDB等数据库,以及AlphaFoldDB等预测结构数据库,呈现出日益丰富和多元化发展态势(见图 1)。这些数据库的建立为科学家进行生物大分子结构与功能研究提供了核心数据,并逐渐成为支撑整个生命科学研究的基石。
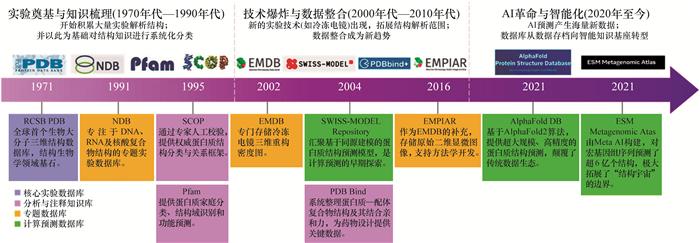
1.1 PDB:结构生物学的基石
PDB数据库是全球首个、也是最重要的生物大分子三维结构数据库。它是现代分子生物学研究不可或缺的基础设施之一,提供的高质量数据为生物大分子结构分析和功能注释奠定了基础,并在近年成为了引爆生物大分子结构AI预测革命不可或缺的“数据燃料”。
1.1.1 历史与发展
PDB成立于1971年,其历史起点仅为7条通过X射线晶体学解析的蛋白质结构记录[26]。PDB的发展与结构生物学实验技术的进步紧密相连,其数据规模和种类持续增长,数据来源逐渐扩展到核磁共振仪和冷冻电镜解析得到的结构。
1.1.2 数据内容与组织结构
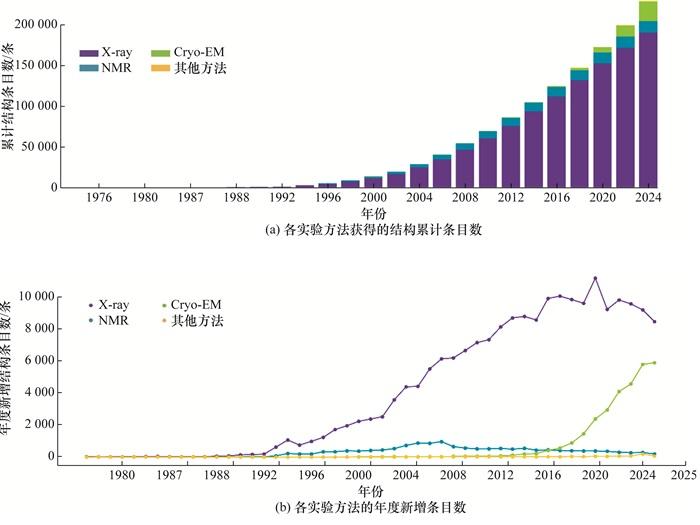
PDB的核心价值在于其存储的高度结构化、经过专家审校的标准化生物大分子结构数据。截至2025年9月,PDB已收录超过24万条结构条目(Entry),涵盖了蛋白质、核酸及其复合物、糖等多种生物大分子,其中约85%条目来自X射线晶体学数据,但冷冻电镜数据增长速度已经超过X射线晶体学数据(见图 2)。
每一个PDB条目都是一个信息丰富的多层次集合,其核心是通过标准且扩展性强的mmCIF格式精确定义了结构中每个原子的空间位置(x、y、z坐标)[31]。此外,PDB还归档了关键的实验原始数据,例如X射线晶体学研究中的结构因子文件和冷冻电镜研究的三维密度图。最后,所有数据都含有丰富的生物学与化学注释,包括分子名称、来源物种、氨基酸或核酸序列,以及结合配体、离子或化学修饰的信息。同时,每个条目都链接到描述其解析过程的原始研究论文,构成了完整的知识追溯链条。这种高度结构化、标准化的数据组织方式不仅保证了科研的严谨性和可重复性,也为下游的数据分析和计算建模提供了坚实基础。
1.1.3 核心功能与使用方法
PDB通过其官方网站(rcsb.org)提供全面的数据服务,包括数据检索、序列搜索、可视化分析、结构比对和数据下载等。在数据检索方面,用户不仅可以通过PDB ID、分子名称或作者等关键词进行快速搜索,还可以利用高级搜索功能构建复杂的查询逻辑,精确筛选出符合特定条件的结构数据集。此外,平台还支持序列搜索,允许用户提交蛋白质或核酸序列,通过BLAST等算法在整个数据库中寻找具有同源序列的已知结构[32]。
1.2 超越坐标:结构分析功能注释数据库
PDB提供了海量的生物大分子原子坐标结构数据,但坐标本身并不能自动揭示其背后的生物学意义。为了将这些原始结构数据转化为可理解的知识,一系列结构分析和以结构为基础的功能注释数据库应运而生。它们通过对结构进行分类、解读和关联,共同构建了一座从三维构象通往生物学功能的桥梁,并为AI时代的计算方法提供了不可或缺的生物学先验和语义约束。
1.2.1 结构分析数据库
结构分析数据库的核心功能,是在原始坐标的基础上,从进化、拓扑和物理相互作用等更高维度对结构数据进行抽象和解读,使得研究者能够从纷繁复杂的构象中发现新的生物机制和原理。
在宏观分类之外,另一类重要的结构分析数据库则专注于对结构进行精细化特征分析与高阶结构解读。例如,以MolProbity为代表的工具集通过检查原子间的碰撞、键长键角和主侧链构象,对结构模型的物理化学合理性进行评估[40]。PDBsum将复杂的蛋白内部及蛋白—配体相互作用网络转化为直观的2D示意图,降低了解读门槛[41]。而PISA(proteins, interfaces, structures and assemblies)则通过分析晶体学接触来推断生物学意义上的蛋白质复合物(即四级结构)[42]。这些数据库将原始坐标加工为具有明确物理或生物学意义的特征,为AI模型提供了关键的物理边界约束和相互作用模式信息。
1.2.2 功能注释数据库
如果说结构分析回答了“它是什么样”,那么功能注释则回答了“它能做什么”。这类数据库致力于建立从序列、结构到生物学功能的可靠关联,最后通过整合将分散的信息编织成一个统一的知识网络。
功能注释的基石是功能模块的识别。在这方面,Pfam数据库是当之无愧的核心工具[43]。Pfam由欧洲生物信息学研究所(EBI)于1997年创建,现由UniProt联盟维护,其关键方法是为每一个蛋白质家族构建一个基于多序列比对的隐Markov模型(hidden Markov model, HMM)[44],并由此从海量未知序列中快速、精准地识别出功能域的边界。截至2024年的35.0版本,Pfam数据库已定义超过19 000个蛋白质家族与功能域[45]。另外,作为补充,SMART同样采用HMM模型,但更侧重于对信号传导、核内及胞外蛋白中可移动功能域的识别[46-47]。Pfam和SMART这样的数据库不仅帮助研究者推断未知序列的生物学作用,也为AI结构预测提供了强大的功能约束,显著提升了结构预测结果的生物学合理性。
然而,关键功能往往由结构中少数关键残基决定。因此,更精细的功能位点注释至关重要。CSA(catalytic site atlas)数据库通过文献挖掘和同源推断,精确标注了酶活性中心的催化残基[48]。这类信息为AI模型提供了最直接的功能约束,指导其生成具有生物学活性的正确构象。
1.3 专题结构数据库:新方法与新焦点的产物
除PDB等通用型数据库外,针对特定技术平台或分子类型的存储需求,还衍生出一批专题结构数据库。这类数据库在元数据组织和下游应用场景上具有高度专门化特征,共同构成了结构数据生态的有机延伸。
1.3.1 冷冻电镜数据资源
冷冻电镜是一项革命性的结构生物学技术,它通过将生物大分子速冻在玻璃态冰中,利用透射电子显微镜获得图像后计算进行结构解析[55]。该技术尤其擅长解析传统晶体学难以处理的、柔性巨大的超大分子复合物及膜蛋白,已成为结构生物学的主流方法之一。
1.3.2 特定生物问题专题数据库
除了技术驱动的数据库,另一类专题库则聚焦于特定的生物分子家族或现象,提供远超通用数据库的深度注释。它们大致可分为2类:一类专注于特定的生物大分子类别,另一类则专注于特定的结构特征或状态。
第1类专题库的一个典型代表是GPCRdb,它专注于G蛋白偶联受体(GPCRs)这一最大的药物靶点家族,在结构数据之外,还提供了突变效应信息,极大地便利了GPCR的药物设计与功能研究[59-62]。同样功能的还有IMGT(international ImMunoGeneTics information system)。作为免疫学领域的黄金标准数据库,它专门收集免疫球蛋白(抗体)、T细胞受体(TCR)等免疫系统相关分子的基因、序列和结构[63-65]。IMGT精确定义了互补决定区(CDR)的边界,这对于抗体药物的研发至关重要。另外一个很典型的代表是针对核酸的NDB (nucleic acid database)是核心资源,它专门归档通过X射线晶体学、核磁共振等方法解析的DNA和RNA三维结构[66-68]。
1.3.3 蛋白质结构预测数据库
近年来,以AlphaFold2为代表的深度学习模型取得了历史性突破,其预测精度在许多情况下已接近实验水平。这一革命性进展以其前所未有的规模和准确性,开启了构建超大规模、全蛋白质组级别结构预测数据库的新纪元。
AlphaFold DB(AlphaFold protein structure database)是这一新纪元的开创者。它由DeepMind与EBI合作建立,2021年首次上线,核心数据是AlphaFold2基于UniProtKB蛋白质序列预测的三维结构[22, 77]。目前该数据库已覆盖人类及数十个物种、数千万条预测结构,每条条目均提供预测的三维坐标文件、基于pLDDT分数的置信度指标,以及与已知实验结构的比对信息[78]。与此相辅相成的是由Meta AI团队发布的基于进化尺度模型的宏基因组图谱(ESM metagenomic atlas),它利用ESMFold模型,对来自环境样本的宏基因组序列进行了超过6亿个结构的预测,致力于探索广阔的“蛋白质暗物质”[79]。
AI预测数据库的出现具有里程碑意义。首先,它们极大地填补了PDB中因实验难度大而缺失的结构空白(如膜蛋白、无序蛋白)。其次,它们将AI的预测结果本身体系化为可检索、可利用的公共资源,形成“AI预测→数据化存储→反哺AI研究”的闭环。这不仅为功能注释、药物发现等下游任务提供了前所未有的数据覆盖,也为后续AI模型的开发提供了新的基准和起点,是AI反哺结构生物学数据生态的典型案例。
1.4 小结
表 1 结构生物信息学常用数据库分类汇总 |
| 数据库类型 | 名称 | 主要数据类型 | 功能定位 | 应用场景 |
| 通用库 | PDB | 蛋白质、核酸及复合物的三维坐标、实验参数文件、密度图等数据 | 全球最大最权威的标准化三维结构数据库,AI训练的黄金数据 | 基础研究、结构生物学、AI模型训练 |
| EMDB | 冷冻电镜三维电子密度图及元数据 | 专注于冷冻电镜解析结果,补充PDB | 结构解析算法开发、冷冻电镜方法学开发 | |
| EMPIAR | 原始二维显微图像及关键中间处理文件 | 作为EMDB的补充资源 | 冷冻电镜结构解析相关算法开发 | |
| 分类库 | SCOP | 蛋白家族、结构域等分类数据 | 蛋白质结构域分类权威数据库 | 蛋白质的结构与进化关系、辅助蛋白质结构预测 |
| CATH | 结构分类、结构域边界注释、功能、进化等数据 | 蛋白质三维结构进行分层分类的权威资源 | 蛋白质结构预测辅助 | |
| 注释库 | Pfam | 多序列比对和隐马尔可夫注释数据 | 标注蛋白质家族、结构域和功能位点 | 结构域注释与功能预测、进化与结构域演化研究 |
| SMART | 蛋白结构域与功能位点数据 | 信号传导、核内及胞外蛋白中可移动功能域的识别 | 推断未知序列的生物学作用、引导AI结构模型预测 | |
| CSA | 文献挖掘和同源推断酶相关活性中心数据 | 标注酶活性中心特征 | 酶工程化设计 | |
| PDBbind | PDB中的蛋白质—小分子复合物的结构以及对应亲和力数据 | 分子相互作用领域关键数据库 | 药物设计和功能AI预测模型 | |
| STRING | 整合结构信息在内的多源数据 | 蛋白质—蛋白质相互作用网络 | 蛋白质在细胞通路和功能模块中的协作关系 | |
| InterPro | 来自Pfam、CSA等多个成员数据库的功能标签签名 | 功能注释的元数据库 | 结构—功能关联分析、蛋白质结构精确预测 | |
| 综合库 | UniProt | 序列、结构、功能域、物种信息、文献引用等海量异构数据 | 最核心的综合性知识库 | 提供生物学研究的各种信息 |
| 专题库 | GPCRdb | GPCR家族蛋白序列、结构、突变、配体等相关数据 | GPCR蛋白家族权威数据库 | 促进GPCR的药物设计与功能研究 |
| IMGT | 免疫系统相关分子的基因、序列和结构数据 | 免疫学领域的黄金标准数据库 | 支持加速抗体药物的研发 | |
| NDB | 核酸相关的实验结构数据 | 特定收录核酸结构数据 | 核酸领域基础研究、核酸疫苗和药物研发 | |
| DisProt | 人工文献检索注释和实验验证的内在无序蛋白数据 | 内在无序蛋白相关的数据 | 内在无序蛋白质的功能研究以及靶向药物研发 | |
| PDBTM | 跨膜蛋白结构和注释 | 跨膜蛋白在膜上的空间排布 | 膜蛋白其通道、转运或信号传导功能 | |
| OPM | 膜蛋白在膜上的空间取向 | 跨膜螺旋的预测和定位 | 膜蛋白相关研究 | |
| 预测库 | SWISS-MODEL | 序列比对以及同源模建的结构数据 | 高精度同源模建数据库 | 实验未知结构蛋白建模 |
| AlphaFoldDB | AlphaFold预测的生物大分子三维结构数据 | 覆盖人类基因组水平的蛋白结构数据库 | 蛋白分类和注释、结构预测模型训练 |
综上所述,当代生物大分子结构数据库已形成一个层次分明、功能互补的有机整体。以PDB为代表的通用库是毋庸置疑的基石;SCOP、CATH、Pfam等分类库和注释库将原始数据转化为知识;EMDB、GPCRdb等专题库满足了方法与学科的深度需求;而AlphaFoldDB等预测库则标志着AI对数据生态的革命性拓展。理解这一分类体系,是有效利用这些资源并洞察其与AI共生关系的基础。
2 生物大分子结构数据库与人工智能的共生关系
作为生命科学的AI革命代表的蛋白质结构预测,建立在整个生物大分子结构数据体系基础之上。然而,AI与数据库之间并非简单的“使用与被使用”关系,而正在逐渐形成一种深度互馈、自我迭代的共生关系。这一关系中的3种关键作用模式包括:核心数据资源对模型训练的奠基性支撑,结构与功能知识对模型推理的引导性增强,AI预测结果对数据库体系的反哺式扩展(见图 3)。这3种模式共同构成了一个强大的“数据-AI-知识”赋能闭环,该闭环正在重塑结构生物学的研究范式。
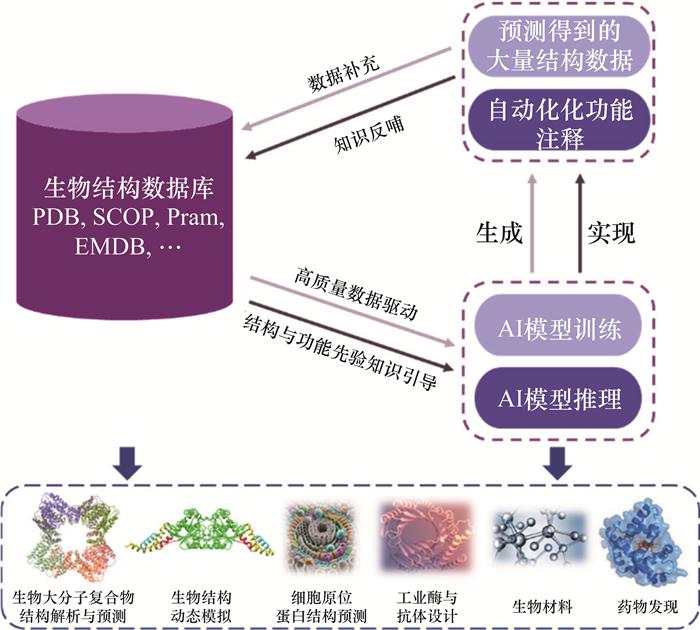
2.1 PDB的结构数据是AI模型训练的基石与燃料
任何成功的AI模型都建立在高质量、大规模的训练数据之上,蛋白质结构预测的AI模型更是如此。正是PDB等数据库数十年如一日积累的高质量实验结构数据,为AI模型提供了学习生物大分子折叠物理规律的基石和燃料。
以AlphaFold2的成功为例,其卓越性能的根基正是对海量异构数据的深度整合与学习。它的训练直接利用了PDB中约17万个实验解析的高质量蛋白质结构作为“答案”,并通过整合蛋白质序列数据库的数亿条序列,使得模型能够同时捕捉到物理化学上的空间约束和进化过程中的保守信号,从而实现了接近实验精度的结构预测。这充分说明,数据库提供的结构信息是模型训练不可或缺的基石。
2.2 数据库的结构功能知识对AI模型训练和推理的增强
PDB等数据库提供的结构数据为模型训练提供了基础,而相关的功能注释数据则提供了更高层次、经过抽象的“知识”。这些知识通过引入生物学先验(biological priors)和提供精确的监督信号(supervisory signals),在AI模型训练及应用中发挥着关键的增强作用。
例如,SCOP数据库提供的结构分类体系为AI训练和推理提供了进化层面的深层约束。该数据库基于专家审核的结构分类信息,能够在模型预测置信度较低的区域辅助推断合理的二级结构排列方式,有效减少因随机采样导致的构象偏差,提升预测稳定性。Pfam数据库的功能域注释体系为AI预测提供了关键的语义约束。其基于HMM构建的系统性功能域分类可直接指导模板选择策略。通过将目标序列与已知功能域进行匹配,AI模型能够优先选择功能相关的模板结构,显著减少盲目搜索的计算开销,同时提高结果的生物学合理性。
2.3 AI模型预测结果对生物大分子结构数据库的反哺、扩展与重塑
AI产生的高置信度预测结构正在成为生物数据库体系的重要组成部分,标志着“数据-AI-数据”闭环生态的形成。这一机制不仅扩展了数据库的规模和多样性,也持续提升了数据库的功能性。
更进一步,预测结构经数据库集成与发布后,可直接为实验研究提供假设来源与设计指导,而实验验证的结果又反馈用于优化AI模型参数与更新数据库内容,从而形成“预测—验证—更新”的持续迭代闭环。这一过程推动了结构生物学数据体系从静态存储库向动态知识系统的根本性转变,体现了AI与数据库融合生态的自我完善能力和持续发展潜力。
3 挑战与局限性
生物大分子结构数据库与AI的融合虽展现出巨大潜力,但其协同发展仍面临多重挑战,主要体现在2个层面:一方面,数据库的内在局限制约了AI模型的能力边界;另一方面,AI的颠覆性力量也对传统的数据库生态提出了严峻的新挑战。更为关键的是,这2个层面的问题并非孤立,而是会相互交织、形成负向循环(见图 4)。全面审视这2个层面,对于指引该领域的未来发展至关重要。
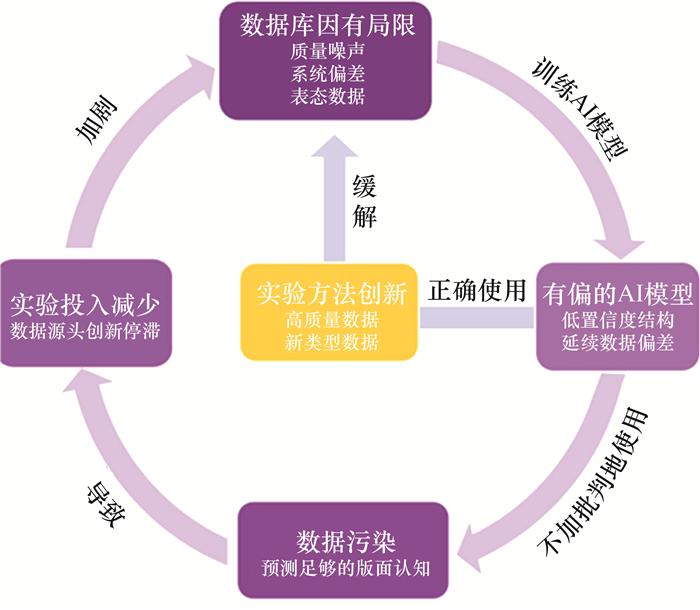
3.1 数据库固有局限性对AI模型的制约
当前AI模型在结构生物学中的性能上限,根本上受限于其训练数据的质量与多样性。数据库的内在缺陷通过训练过程被传递并放大,成为AI结构预测能力的核心瓶颈。
进一步而言,数据库的静态特性限制了AI对生物动态过程的理解。现有条目主要记录生物大分子在特定条件下的静态构象,缺乏对其在细胞内动态组装、变构调节等过程的表征。数据库中复合物结构和构象变化数据的缺失,使AI模型难以学习蛋白质的变构效应、柔性区域动态等关键生物学特征[98]。此外,几乎所有结构数据均来自高度纯化的体外条件,与拥挤复杂的细胞内环境相去甚远,基于这些“理想化”数据所做的预测在生理情境下的有效性仍需审慎评估。
最后,也许对未来影响更深远的一项局限是数据库内容的进化惯性(evolutionary inertia)。PDB中的结构绝大多数是自然界亿万年进化而来的天然蛋白质的野生型结构,缺乏突变体结构以及非自然选择的、理论中可能存在的新结构。这一局限导致AI模型在预测疾病相关蛋白,以及在蛋白质从头设计新结构、新功能开发方面的表现欠佳。AI被训练成了自然进化的“模仿者”,而非一个能探索“结构可能性空间”的创造者,这严重限制了其在精准医疗和合成生物学中的前沿应用。
3.2 AI对数据库生态带来的新挑战
AI技术的快速发展在带来突破、拓展结构生物学研究领域的同时,也给传统生物大分子结构数据库生态带来了数据质量、基础设施,乃至学科发展等方面的全新挑战。
首先,最直接的挑战来自AI预测结果可能引发的“数据污染”问题[99]。AlphaFoldDB等数据库含有数亿级别的预测结构,它们本质上是基于现有知识的计算推断或“假说”,而非经过实验验证的“事实”。若不加批判地使用这些质量参差不齐的预测数据,可能对整个知识生态系统造成污染,导致研究者基于低置信度预测设计实验,造成资源浪费并影响科研严谨性。
再次,更深层次的担忧在于AI的成功可能导致实验结构生物学的创新停滞。若形成“预测已经足够好”的认知,可能会削弱对冷冻电镜、X射线晶体学等实验技术持续创新的投入意愿。然而,AI模型的进步恰恰依赖更多样、更高质量的实验数据作为训练基础,以突破其当前在预测复合物、动态和全新折叠类型等方面的瓶颈。如果数据源头停滞,AI自身的发展终将成为无源之水,整个领域可能陷入低水平的自我循环[103]。
最后,AI产出对数据库基础设施提出前所未有的需求。存储和维护数亿级别的预测结构,并提供高效的检索、比对和版本控制,对计算和存储资源带来巨大压力。如何将实验数据与预测数据进行有效整合与区分展示,如何建立AI原生(AI-Native)的数据库架构以适应模型的快速迭代,是传统数据库管理者面临的全新课题[104]。
4 未来展望:迈向人工智能原生的生物大分子结构基础设施
AI与生物大分子结构数据库的协同发展,正在推动“预测—生成—验证—应用”的全链条研究范式转变。为适应这一趋势,未来数据库将逐步从被动存储库演进为支持AI深度集成的新型知识基础设施,这一转变远非简单的功能升级,而是涉及数据库架构、功能及其在科研流程中角色的系统性重构。这一演进遵循着清晰的技术路径:数据库架构将沿“AI增强→深度集成→AI原生”的轨迹进化;同时,科学研究范式也将发生三大深刻转向——从单一蛋白到复合体系、从孤立分子到原位环境、从理解自然到全新设计;其最终目标是构建承载“数字孪生”的可信、共享动态知识引擎(见图 5)。
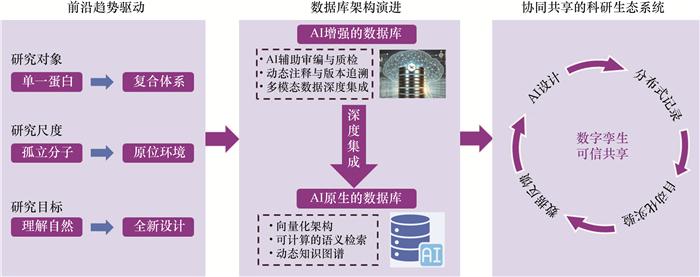
4.1 前沿趋势驱动的数据库发展需求
未来数据库的发展方向主要由三大科学前沿趋势所驱动,这些趋势对数据内容、标准和处理能力提出了新的要求。
从研究对象维度看,重点正从单一蛋白质向多分子复合体系拓展[105]。生命过程依赖于蛋白质、核酸和聚糖等生物大分子的协同作用。然而当前数据库中非蛋白质分子的结构数据严重不足。随着AlphaFold3等新一代模型展现出预测蛋白质-核酸相互作用及翻译后修饰的能力,数据库需要建立新的标准和注释规范,以表征这些化学性质和拓扑结构更多样化的分子,并能清晰描述它们之间复杂的相互作用网络。
从研究尺度维度看,范围正从孤立分子向细胞环境下的原位结构生物学延伸。冷冻电子断层扫描(Cryo-ET)等技术提供了分子在接近生理环境中的结构信息[106],未来目标是通过整合多尺度原位数据,构建细胞器乃至整个细胞的动态结构模型[107]。这要求数据库具备存储和处理原位成像数据、亚细胞定位信息及分子动态轨迹的能力,为AI理解生理环境中的结构功能提供数据基础。从研究目标维度看,方向正从理解自然结构转向设计全新功能分子。以RFdiffusion等为代表的生成式AI能够创建自然界中不存在的蛋白质,产生大量具有潜在应用价值的全新设计结构[108]。这对数据库提出了新挑战:需要建立有效机制来区分、存储和标注这些人工设计分子,并将其与实验验证的功能数据进行关联,构建服务于合成生物学与蛋白质工程的“设计-结构-功能”知识库。
4.2 从智能增强到AI集成的架构演进
为应对上述需求,数据库架构将经历从智能增强到深度集成的渐进式演进过程,最终形成AI原生的新一代基础设施。
在可预见的未来,PDB等生物大分子结构数据库仍将以结构实验数据为基石,并通过AI技术,来增强现有功能。针对AI预测结果和实验数据的爆炸式增长,可以引入AI辅助的数据审编流程,在新结构提交时自动检查其格式及理化属性等。另外,注释系统将实现动态更新与版本追溯,明确标注预测结构的来源模型版本,并在新模型发布后自动更新或并列展示预测结果,形成可比较的演进记录。
同时,数据整合方式也将从简单的ID链接转向内容层面的深度融合。数据库将利用AI技术自动识别并关联PDB结构域、EMDB密度图和Pfam功能注释,在三维空间上进行对齐和交叉验证,为研究者提供预处理好的、可靠的知识视图,而非需要用户自行清洗的原始数据。
4.3 构建协同共享的科研新生态
生物大分子结构数据库的发展将推动形成更加开放、协同的研究生态系统,通过数据共享和技术集成加速科学发现进程。
例如,可以用生物大分子结构为中心,深度整合多模态数据,为关键生物实体构建“数字孪生”(digital twin)[111]。一个蛋白质的“数字孪生”将是一个动态的、聚合性的知识中心,包括其序列、实验和预测结构、构象动态数据、原位成像信息、相互作用网络及功能数据等。这种全景式数据形式将为AI提供前所未有的学习素材。
更重要的是,生物大分子结构数据库将成为连接计算与实验的自动化闭环核心,并可借助分布式账本(如区块链)等技术提供信任保障。一个典型的未来工作流可能包括:通过生成式AI设计新型酶分子,设计方案被存入数据库并通过区块链技术记录来源和时间戳;系统自动生成实验验证任务并通过API发送至自动化实验平台;实验结果自动回传并与原始方案关联并同样被记录在区块链上。这种机制不仅能确保研究过程的透明与可信,还能通过智能合约建立去中心化的激励机制,鼓励研究者共享高质量实验数据,促进全球范围内的开放科学合作。
通过这种转变,数据库将从记录历史的知识仓库演进为驱动未来发现的创新引擎。它不仅保存已有知识,更主动参与知识的创造与验证,最终构建一个更加开放、可信和高效的全球协同研究新生态。
5 结论
本文系统介绍了生物大分子结构数据库历史演进与现状,着重阐述其与AI技术之间日益紧密的协同关系,以及这一关系对结构生物学研究范式产生的深远影响,并在此基础上展望其可能的未来发展。从PDB等核心数据库的建立,到SCOP、Pfam等功能注释数据库的发展,再到AlphaFoldDB等预测数据库的出现,不断丰富、扩展的数据生态为AI模型提供了至关重要的数据基础和学习素材,而AI的预测结果又反过来扩展数据库的规模和覆盖范围,形成“结构数据库-AI模型”双螺旋上升的闭环,推动结构生物学从传统的实验驱动向计算与实验深度融合的新范式转变。
然而,这一协同发展仍面临重要挑战。数据库中存在的数据偏差、质量不均、动态信息缺失、进化惯性等内在局限,以及AI技术带来的预测结果可靠性、数据污染风险、数据治理等新挑战,都需要通过技术创新和数据完善来系统解决。
未来,构建AI原生的新一代生物大分子结构知识基础设施将成为关键发展方向。这一进程将经历从智能增强到深度集成的渐进式演进。近期通过AI辅助数据审编和多模态整合提升数据质量与可用性;远期则向深度向量化的AI原生架构转变,使数据库成为可计算的智能底座。最终,数据库将不仅是知识的存储库,更是生物大分子结构“数字孪生”的载体及驱动科学发现的智能平台,从而推动结构生物学从理解生命结构向理性设计生命功能的新阶段发展。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}